
ClinicalTrials.gov Educational Opportunity
ClinicalTrials.gov: Essentials for Academic Medical Centers
This virtual training from FDA and NIH specialists is designed to help academic researchers and their staff meet the federal requirements for registering clinical studies and reporting results on ClinicalTrials.gov.
The live virtual sessions will be held on:
July 14 at 10:00 am
July 22 at 10:00 am
July 30 at 10:00 am
What is ClinicalTrials.gov?
ClinicalTrials.gov is a resource that provides access to information on clinical trials studying a wide range of diseases, conditions and interventions. Studies listed in the database are conducted in all 50 States and worldwide. Each ClinicalTrials.gov record includes summary information about studies.
Federal regulations require registration and results reporting on ClinicalTrials.gov for applicable clinical trials (ACTs) under FDAAA 801 and / or per NIH Policy on the Dissemination of NIH-Funded Clinical Trial Information. Applicable trials must be registered on clinicaltrials.gov before participant enrollment begins.
The risk of noncompliance with these requirements includes civil monetary penalties in excess of $15,000 per day, civil or criminal actions, and/or loss of NIH funding for the institution.
Problems with CT.gov Record?
Want a quick start guide to CT.gov?
Registration Requirements
ClinicalTrials.gov Registration Decision Tool
Use the decision tool to determine if registration on CT.gov is required
Registration Requirements:
NIH funded clinical trials are required to be registered on clinicaltrials.gov:
Per NIH Policy on the Dissemination of NIH-Funded Clinical Trial Information - Effective January 18, 2017, if your study is NIH funded and meets the NIH definition of a clinical trial, then clinicaltrials.gov registration is required.
See NIH Decision Tool!
- For NIH funded research, use the following four questions to determine the difference between a clinical study and a clinical trial:
- Does the study involve human participants?
- Are the participants prospectively assigned to one or more interventions?
- Is the study designed to evaluate the effect of the intervention on the participants?
- Is the effect being evaluated a health-related biomedical or behavioral outcome?
- If the answers to ALL 4 questions are “yes”, your study meets the NIH definition of a clinical trial.
- Clinical trials subject to the regulation are generally called "Applicable Clinical Trials."
- Applicable Clinical Trials or ACTs are required to be registered on ClinicalTrials.gov not later than 21 calendar days after the enrollment of the first participant.
Basic Experimental Studies Involving Humans (BESH)
For applications submitted for due dates on or after May 25, 2026, BESH are no longer considered clinical trials by the NIH. Therefore, BESH will not have to follow the requirements for clinical trials, including registration and reporting in ClinicalTrials.gov (see NOT-OD-26-032).
-
Applications submitted before May 24, 2026, BESH will continue to be treated as clinical trials including registration and results submission on clinicaltrials.gov.
-
Currently funded BESH studies should continue to follow the established terms and conditions of their respective grant or contract award and guidance as noted in NOT-OD-25-134.
-
See NOT-OD-26-067 for a table of notable changes between applications proposing BESH submitted on or before May 24, 2026 and on or after May 25, 2026.
Harmonizing the NIH Definition of “Intervention” with the Common Rule:
NOT-OD-26-063 – Effective March 27, 2026
OLD NIH definition of an intervention:
-
A manipulation of the subject or subject’s environment for the purpose of modifying one or more health-related biomedical or behavioral processes and/or endpoints.
NEW definition in line with the 2018 Common Rule (45 CFR 46, Subpart A [Protection of Human Subjects]), which defined an intervention as:
-
Both physical procedures by which information or biospecimens are gathered (e.g., venipuncture) and manipulations of the subject or the subject's environment that are performed for research purposes.
NIH Notice of award:
The NIH Award Notice includes a "Clinical Trial Indicator" in Section IV which specifies whether the award supports any NIH defined clinical trials and will specify whether the study is required to be registered. See this example
")
Registration is required for studies that meet the definition of an "applicable clinical trial" (ACT) and either were initiated after September 27, 2007, or initiated on or before that date and were still ongoing as of December 26, 2007.
ACTs, as defined in section 402(j) of the PHS Act, include the following:
- Controlled clinical investigations (other than phase 1 investigations) of any U.S. Food and Drug Administration (FDA)-regulated drug or biological product for any disease or condition
- Certain studies of FDA-regulated medical devices, excluding small clinical trials to determine feasibility and certain clinical trials to test prototype devices, but including FDA-required pediatric postmarket surveillances of a device product
Resource:
ACT Checklist: For complete statutory definitions and more information on the meaning of Applicable Clinical Trial, see the Checklist and Elaboration for Evaluating Whether a Clinical Trial or Study is an Applicable Clinical Trial (ACT)
References:

ACT – important definitions:
Is the study interventional (a clinical trial)?
- Participants are prospectively assigned to an intervention(s) to evaluate the effect of the intervention(s) on biomedical or other health-related outcomes. [Source: 42 CFR 11.10(a); 81 FR 65140-41]
Does the study evaluate* at least one U.S. FDA-regulated drug, biological, or device product?
FDA-regulated Device Product means:
- A device product subject to (1) a finding of substantial equivalence under section 510(k) of the FD&C Act, (2) under section 515 - requiring a premarket approval application (PMA) for the device product, or (3) a marketing application for a Humanitarian Use Device (HUD) - a Humanitarian Device Exemption under section 520(m) of the FD&C Act.
- Device products that are considered to be subject to section 510(k), 515, or 520(m) of the FD&C Act include significant risk devices (SR) for which approval of an IDE is required, non-significant risk devices (NSR) that subject to abbreviated IDE requirements (21 CFR 812.2(b)), or device products that are exempt from the submission requirements (IDE Exempt) of 21 CFR part 812.
- It DOES NOT include device feasibility, Phase 0 or Phase 1 trials.
*The device is under clinical investigation and is the object of the investigation.
FDA-regulated Drug Product means:
- A drug that is the subject of an approved NDA (new drug application) or BLA (biologic license application) or that would require an approved NDA or BLA to be legally marketed in the United States.
- A clinical trial including an intervention with a “dietary supplement” could be an ACT.
- It DOES NOT include Phase 0 or Phase 1 drug / biologic trials. For determining whether a drug / biological is the variable of interest, see page 8 of the ACT checklist
Registration Requirements per International Committee of Medical Journal Editors (ICMJE):
- In 2005, the ICMJE defined trials that must be registered in order to be considered for publication in journals that adhere to ICMJE standards.
- The ICMJE requires registration of clinical trials in a public trials registry at or before enrollment of the first participant to be considered for publication. Which trials registries are acceptable to the ICMJE?
- Which journals are members of the ICMJE? The editors of 17 journals are currently official members of the ICMJE.
- Many Journals (not limited to medical journals) have adopted the registration policy. We recommend you contact the Journal directly to see if they follow ICMJE Recommendations.

The ICMJE definition of clinical trial: "any research study that prospectively assigns human participants or groups of humans to one or more health-related interventions to evaluate the effects on health outcomes.”
- With or without concurrent comparison or control groups,
- Health-related interventions are those used to modify a biomedical or health-related outcome; examples include drugs, surgical procedures, devices, behavioral treatments, educational programs, dietary interventions, quality improvement interventions, and process-of-care changes.
- Health outcomes are any biomedical or health-related measures obtained in patients or participants, including pharmacokinetic measures and adverse events.
- See ICMJE Clinical Trials Registration website for details.
- Effective January 1, 2014, the Centers for Medicare and Medicaid Service (CMS) require the mandatory reporting of the ClinicalTrials.gov Identifier (NCT Number) number on claims for items and services provided in clinical trials that are qualified for coverage under the Medicare National Coverage Determination (NCD) Manual, Section 310.1
- Any claim that does not include the NCT number will be returned to the billing provider and may not be paid. If the study has been determined to be a Qualifying Clinical Trial (QCT) under Medicare’s NCD and the investigator intends to enroll Medicare beneficiaries, the study must be registered on ClinicalTrials.gov regardless if it meets the criteria for registration under the FDAAA, NIH and/or ICMJE requirements.

Department of Defense
Please check with your Program Officer if registration is required.

Patient-Centered Outcomes Research Institute (PCORI)
- Please review PCORI’s Process for Peer-Review of Primary Research and Public Release of Research Findings
- If your study qualifies as a clinical trial, registration is required prior to enrollment of the first patient.

NIH National Cancer Institute
- NEW RESOURCE: NCI Registration and Reporting Requirements Presentation
- All NIH-funded investigators including NCI investigators are subject to the NIH policy.
- “Covered Trials” means all initiated or commenced NCI-Supported Interventional Clinical Trials whether extramural or intramural.
- For every Covered Trial, Final Trial Results are expected to be reported in a publicly accessible manner within twelve (12) months of the Trial’s Primary Completion Date regardless of whether the clinical trial was completed as planned or terminated earlier.
To comply with the Policy, Final Trial Results may be reported in a publicly accessibly manner in various ways, which include but are not limited to ClinicalTrials.gov.

Department of Veterans Affairs
- Principal Investigators (PIs) of VHA Office of Research and Development (ORD) funded clinical trials are responsible for registering their trials with and submitting summary results to ClinicalTrials.gov, as a condition of funding.
- ORD uses the same definition of a clinical trial as the World Health Organization / ICMJE. This definition is "any research study that prospectively assigns human participants or groups of humans to one or more health-related interventions to evaluate the effects on health outcomes."
If you have a clinical trial that includes a drug that's available via expanded access:
Per FDA Guidance for Industry if the party responsible for registering that clinical trial is not both the sponsor of the applicable trial and the manufacturer of the investigational drug product being studied, that responsible party is not required to submit information on the availability of its investigational drug product for expanded access. For further information, refer to the “Frequently Asked Questions” section on the ClinicalTrials.gov website.
See: Expanded Access Information and Submission Deadlines
A physician who submits an individual patient expanded access Investigational New Drug Application (IND), including for emergency use, as specified in 21 CFR 312.310, to the U.S. Food and Drug Administration generally would not be required to submit expanded access information to ClinicalTrials.gov.
How to Register & Update the Record
How to Register & Update the Record
The following user guide explains how to carry out some of the most common functions on ClinicalTrials.gov when registering a study. For a general overview of the registration process and requirements, see How to Register Your Study.
Protocol Registration and Results System (PRS) Guidance
EQUIP TIP: Submit to the IRB prior to releasing the clinicaltrials.gov record for PRS review
Once you have determined that your study needs to be registered on ClinicalTrials.gov, you will need to submit the record for the study in the Protocol Registration and Results System (PRS):
- For PRS access (if you do not already have an account), please contact Electronic Research Administration: era@research.uci.edu.
- If you are unsure if your study should be registered, contact your PRS Administrator:
- For UCI Health Science studies, please contact Jinah Chang (jinahec@hs.uci.edu )
- For UCI Cancer Center studies, please contact Michelle Tran (mdich@hs.uci.edu)
- For Non-UCI Health studies, please contact the EQUIP team: Anu Mathur, EQUIP manager (anuradhm@uci.edu or Laura Ulloa (ulloal@uci.edu) EQUIP Senior Analyst.
- Once the account has been created, an email will be sent from ClinicalTrials.gov to the new user with a username, password and instructions for logging in to UCI’s Institutional PRS account
- For questions about the registration process at UCI, contact the EQUIP team, or contact PRS staff at register@clinicaltrials.gov.
- Go to https://register.clinicaltrials.gov/ to sign in
- Enter Organization: UCaliforniaIrvine
- Enter the username and password emailed to you by ClinicalTrials.gov

- Change your password once you log in for the first time
- Go to Accounts > Change password
- You will receive a temporary password via email
- Suggestion: Print or save a copy of the Protocol Data Entry Protocol Review Criteria (PDF) for reference. This document provides general guidance for compliant record creation as well as “Hints” detailing specific requirements for completion of key fields in the protocol record.
- On the right side of the Modernized PRS home page, press “Create New Record” to begin a new PRS record.
- Only one Record Owner can be assigned to a study record, but the Record Owner can allow other users to edit the study record by granting them access.
- For UCI Investigator Initiated studies, the Principal Investigator (Lead Researcher) or the responsible Clinical Research Coordinator can be listed as the “Record Owner”.
- Other study staff requiring PRS access should be added to the “Record Access” list via the “People” button on the right side of the main record screen:
To Begin Entry of Information For The Record:
On the main record page, click on the “Protocol” tab:
**EQUIP TIPS TO AVOID COMMON PRS ERRORS:
STUDY DESCRIPTION
- Brief Summary does not unnecessarily duplicate information provided for other data elements
- Brief Summary clearly states the study’s hypothesis or the purpose (for interventional and observational)
- Brief Summary and Detailed Description are written in complete sentences with no formatting errors
- Record does not use personal pronouns:“I, we, our, us, they, them, their” – becomes “the investigator(s)”; “you, your” – becomes “the participant(s)”
CONDITIONS
- Conditions/Focus of study are discrete and does not use verbs or complete sentences
- Keywords are not numbered or bulleted, each condition and keyword is listed individually, one per line
STUDY DESIGN
- All required fields are completed
- Verify Study Design based on protocol in IRB
- “Allocation” marked as “N/A” for single-arm studies
- Enrollment number Actual/Anticipated verified
ARMS/INTERVENTIONS
- Arm Title or Group/Cohort Label is brief and informative (even if there is only 1 arm)
- Interventions and intervention descriptions are entered correctly
- Arms/interventions are cross-referenced appropriately
OUTCOME MEASURES
- Title is specific and states WHAT is being measured
- only 1 variable must be assessed per outcome measure
- Description explains HOW outcome is being measured, not WHY it is being measured
- Scoring scale name, score range, significance of upper and lower limits specified (if applicable)
- Unit of measure specified
Time frame specified as a single time point or change between 2 time points
INCORRECT: “Safety and Toxicity”, Description: “Safety of study drug.”
CORRECT: “Safety as assessed by number of participants experiencing adverse events” Description: “Number of participants experiencing adverse events grade 3 or higher, as defined by Common Terminology Criteria for Adverse Events version 5.0 (CTCAE v5.0)”
ELIGIBILITY
- Age Limits are consistent with the Eligibility Criteria and with other parts of the record
- Eligibility criteria is divided into Inclusion/Exclusion criteria in bulleted format
CONTACTS/LOCATIONS
- Central Contact Person listed as a primary research team contact
- Study Officials: Person responsible for overall scientific leadership of the protocol, including the study Principal Investigator.
- Organization Affiliation: Full name of the Official’s organization (for UCI Researchers, its “University of California Irvine”)
- All study sites specified matches IRB
- Recruiting status for each study site accurate (if at least one study site is recruiting then Study Status reflects “Recruiting”)
- Each facility is listed in a separate field
IPD Sharing Statement
- Field is completed with a ‘Yes’ or ‘No’ selection
- If ‘Yes’ is selected, an IPD Sharing Plan is identified
- The Plan to Share IPD selection is consistent with the IPD Sharing Plan Description.
REFERENCES
- Each citation is listed in a separate field (if applicable)
-
- You will be prompted for Organization’s Unique Protocol ID.
- At UCI, the Unique Protocol ID MUST match the IRB number (DIGITS ONLY no dashes, no #, same number as in Zot IRB).
- Add the brief and official title for your study
- if using any acronyms, specify them in the “Acronym” section
- Select Continue to complete the “Create New Record” module and add information into each module of the protocol record as appropriate to your study.
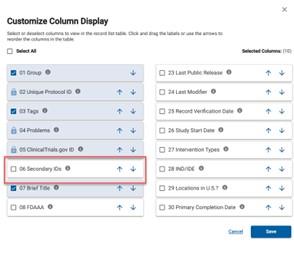
- Secondary IDs:
- Grant‐funded projects MUST enter the sponsor‐issued grant or award number in this field. [1]
- For industry‐funded projects, use the sponsor’s protocol ID number
- For UCI Cancer Center studies, in the Secondary ID field, select "Other Identifier" and add the unique CFCCC identifier, with "Issuing Organization" as "UCI CFCCC".
[1] U.S. National Institutes of Health (NIH) Grant/Contract Award Number: In the Secondary ID field, include activity code, institute code, and 6-digit serial number. Other components of the full award number (type code, support year, and suffix) are optional.
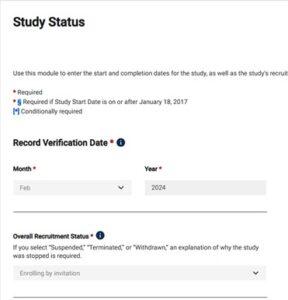
- The Record Verification Date reflects the last time the PRS record was updated. Revise this date each time the record is verified for accuracy and completeness.
- Complete the rest of the required information per guidance in this section:

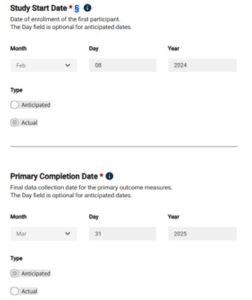
- If unsure of dates, indicate the date as “anticipated” in the last dropdown under “Type”
- If recruitment has begun, specify the “actual” date the first subject was enrolled.
- For all UCI Investigator Initiated studies, the Principal Investigator (PI) should be designated as the Responsible Party (RP).
- Under Responsible Party choose “Principal Investigator”.
- Select the Investigator name from the drop-down menu (email ERA@uci.edu if the PI does not have a PRS account).
- Enter the Investigator’s Official
- Investigator’s Affiliation should automatically populate to “University of California, Irvine”
- Sponsor: Regardless of funding source, enter the “regulatory sponsor” (primary organization overseeing the implementation of the study), usually University of California Irvine.
- Any collaborating sites should be entered under the Collaborators section.
- US. FDA Regulated Drug / Device: Indicate whether this study involves an FDA‐regulated drug, biologic, or device.
- US. FDA Regulated Drug / Device: Indicate whether this study will be conducted with a drug/device product under a U.S. FDA Investigational New Drug (IND) Application or Investigational Device Exemption (IDE).
- Human Subjects Protection Review: Enter the current status of your IRB Protocol in Zot IRB (e.g., “submitted, pending” if an application has been submitted but is under review.
- Add the following information for the UCI IRB:
- Board Name: University of California Irvine IRB
- Board Affiliation: University of California Irvine
- Board Contact: 949-824-8170
- Email: IRB@uci.edu
- Business Address: University of California, Irvine, Office of Research, Irvine, CA 92697-7600
- Data Monitoring: Indicate whether a data monitoring committee (board) has been appointed for this study
- Oversight Authorities: Name each national or international organization with authority over the protocol (e.g. DHHS, FDA, NIH, DOD, DOE, etc.)
**EQUIP TIP TO AVOID COMMON PRS ERRORS:
- IND/IDE information completed (if applicable)
- Enter all the applicable information regarding the study you are registering in the Study Description”, “Conditions”, “Study Design”, “Arms and Interventions”, “Eligibility” sections, as well as any other applicable sections.
- Review and reference the PRS User Guide
- After filling in the last data entry page, the “Protocol Section” page appears with all of the information Review and “Entry Complete” if all information is complete.
- Address any ERROR messages if populated.
**GENERAL EQUIP TIPS TO AVOID COMMON PRS ERRORS:
- Record Verification Date is current Month and year
- Unique Protocol ID is the IRB number ONLY (No other Unique protocol ID’s, IRB number in digits only, no dashes, #)
- Record Owner is the PI or Research Coordinator
- Contact info for Record Owner is up-to-date
- PI on record matches IRB PI
- NCT# included in IRB “Clinical Trials Information” section
- All Warnings/Errors addressed
- All parenthetical citations have been removed
- All acronyms have been expanded on their first use
- Spell-check complete
- Free-text fields are blank if there is no information to report, and do not contain text such as “TBD,” “Pending,” “N/A,” “None”
- If the PI is the responsible party, they will review all entries made by study staff prior to release.
- Use the Record List custom filter to check for records that are Ready for Review and Approval. A system-generated email notification of completed records will also be sent.
- Select Open Record on your Record List.
- Review the record by following steps described in Section 5.1: Data Entry Review.
- Update Record Verification Date to the current month and year.
- Select Approve on the Record Summary page.
- Once released, PRS staff perform final review and processing of the record.
- Note: some records may receive PRS Review Comments that identify major issues that must be addressed by the responsible party before the record will be made available on ClinicalTrials.gov.
- Following successful PRS review, records are made available to the public through the ClinicalTrials.gov web site within 2 to 5 days of Release.
- The ClinicalTrials.gov Identifier (NCT number) is assigned as part of that process.
- Please download and review the IRB EQUIP TIPS guidance document called “CHECKLIST TO ADDRESS COMMON PRS ERRORS” to avoid common issues noted by the ClinicalTrials.gov database (PRS) staff prior to public release.
- Directly email PRS staff if you have any questions or need help related to PRS Review Comments at register@clinicaltrials.gov
- Once a record is created, you can update it as long as it is not undergoing PRS review:
![]()
When / how often must I update ClinicalTrials.gov registration information?
- The Record Verification Date should be updated any time the responsible party reviews the complete set of submitted clinical trial information for accuracy and not less than every 12 months, even if no other updated information is submitted at that time.
- Responsible Parties should review the Clinical Trial Registration Data Elements for More Frequent Updating Table as many elements must be updated within 30 days of a change.
- It is recommended that the Record Verification Date be updated at least every 6 months for studies that are not yet completed, even if there were no changes to the record.
Clinical Trial Registration Data Elements for More Frequent Updating
| Data Element | Deadline for Updating (i.e., not later than the specified date) |
|---|---|
| Study Start Date | 30 calendar days after the first subject is enrolled (if the first human subject was not enrolled at the time of registration). |
| Intervention Name(s) | 30 calendar days after a nonproprietary name is established. |
| Availability of Expanded Access | 30 calendar days after expanded access becomes available (if available after registration); and 30 calendar days after an NCT number is assigned to a newly created expanded access record. [1] |
| Expanded Access Status | 30 calendar days after a change in the availability of expanded access. |
| Expanded Access Type | 30 calendar days after a change in the type(s) of available expanded access. |
| Overall Recruitment Status | 30 calendar days after a change in overall recruitment status. [2] |
| Individual Site Status | 30 calendar days after a change in status of any individual site. |
| Human Subjects Protection Review Board Status | 30 calendar days after a change in status. |
| Primary Completion Date | 30 calendar days after the clinical trial reaches its actual primary completion date. |
| Enrollment | At the time the primary completion date is changed to "actual," the actual number of participants enrolled must be submitted. |
| Study Completion Date | 30 calendar days after the clinical trial reaches its actual study completion date. |
| Responsible Party, by Official Title | 30 calendar days after a change in the responsible party or the official title of the responsible party. |
| Responsible Party Contact Information | 30 calendar days after a change in the responsible party or the contact information for the responsible party. |
| Device Product Not Approved or Cleared by U.S. FDA | 15 calendar days after a change in approval or clearance status has occurred. |
| Record Verification Date | Any time the responsible party reviews the complete set of submitted clinical trial information for accuracy and not less than every 12 months, even if no other updated information is submitted at that time. |
Want to Avoid Common Errors?
Use this checklist!
Reporting Results
Reporting Results
Who is responsible for reporting results?
The Principal Investigator is the Responsible Party that must publish the results on ClinicalTrials.gov.
Walkthrough Webinar
What studies need to report results?
The Food and Drug Administration (FDA) and National Institutes of Health (NIH) require the publication of results for certain studies on ClinicalTrials.gov (Ct.gov).
-
-
- See FDAAA 801 and the Final Rule for more information.
-
The ICMJE Policy recommends results publication but it is not required.
When is results reporting required?
Submission of results information is required no later than 12 months after the Primary Completion Date (the last subject last visit) of the clinical trial, which is defined as the date of final data collection for the primary outcome measure.
How to Submit Your Results?
- See How to Submit Your Results
- Participant Flow. A tabular summary of the progress of participants through each stage of a study, by study arm or comparison group. Participant Flow Data Preparation Checklist (PDF)
- Baseline Characteristics. A tabular summary of the data collected at the beginning of a study for all participants, by study arm or comparison group. Baseline Characteristics Data Preparation Checklist (PDF)
- Outcome Measures and Statistical Analyses. A tabular summary of Outcome measure values, by study arm or comparison group. Outcome Measure Data Preparation Checklist (PDF)
- Adverse Events. A tabular summary of all anticipated and unanticipated Serious adverse event and a tabular summary of anticipated and unanticipated other adverse events exceeding a specific frequency threshold. Adverse Event Data Preparation Checklist (PDF)
- Administrative information consists of the study results point of contact and any agreement between the sponsor and principal investigator (PI) restricting the ability of the PI to discuss the results after the completion of the study.
CHECKLIST TO ADDRESS COMMON PRS ERRORS
Posting the Consent Form
Posting the Consent Form
All clinical trials funded by an agency that has signed onto the Common Rule initially approved on or after January 21, 2019 OR that transitioned to the 2018 Common Rule Requirements (45 CFR 46.101(l)) and the transition determination was documented and dated by the IRB before the trial is closed to recruitment and 60 or fewer days before the last protocol-required study visit by any subject enrolled in the protocol.
OHRP Guidance
Posting the Consent Form
WHAT TO POST
One IRB-approved consent form used to enroll subjects. Even if multiple versions were approved by the IRB, only one must be uploaded.
WHEN TO POST
After the clinical trial is closed to recruitment, and no later than 60 days after the last study visit by any subject, as required by the protocol.
WHERE TO POST
Two federal websites have been identified as satisfying 45 CFR46.116(h): ClinicalTrials.gov and a designated docket folder on Regulations.gov (Docket ID: HHS-OPHS-2018-0021).
ClinicalTrials.gov FAQs
Burden of time: Per DHHS 42 CFR Part 11, the initial submission of registration information will take an average of 8 hours, with each update requiring 2 hours apiece and will take place 8 times during the course of the study (an average of 32 hours!).
DHHS expects that voluntary registrations will:
- submit the same clinical trial registration information as applicable clinical trials.
- expects that required elements of the record are updated as frequently as information for applicable clinical trials.
Voluntary registration places a burden on the Responsible Party to comply with all registration and update requirements. Any ongoing non-compliance with these requirements is on the part of the Responsible Party.
| Name | Type | Intervention Type | Registration Policy Scope | Results Submission Policy Scope |
|---|---|---|---|---|
| Final Rule for Clinical Trials Registration and Results Information Submission (42 CFR Part 11) | U.S. Federal regulation implementing FDAAA 801 effective in 2017 | Drug, biological, and device products | Clinical trials of a Food and Drug Administration (FDA)-regulated drug, biological, or device product other than Phase 1 (drug/biological products) or small feasibility studies (device products) | Same scope as registration |
| NIH Policy on the Dissemination of NIH-funded Clinical Trial Information | National Institutes of Health (NIH) policy effective in 2017 | Any (includes drug, biological, and device products, as well as surgical procedures, and behavioral interventions) | Clinical trials funded in whole or in part by NIH | Same scope as registration |
| International Committee of Medical Journal Editors (ICMJE) Policy | Publication policy initiated by the International Committee of Medical Journal Editors (ICMJE) in 2004 | Any (includes drugs, biological, and device products, as well as surgical procedures, and behavioral treatments) | All interventional studies, including Phase 1 studies; defines criteria for "acceptable registries" | The ICMJE expects authors to meet results information submission requirements "of their funding and regulatory agencies" and "encourages... results reporting even when not required." |
 The “Responsible Party” refers to the entity or individual who is responsible for registering a trial on ClinicalTrials.gov.
The “Responsible Party” refers to the entity or individual who is responsible for registering a trial on ClinicalTrials.gov.- The Responsible Party is responsible for the initial release of the record, all future updates and ensuring the trial registration stays accurate and up-to-date.
- At UCI, the PI serves as the responsible party if they meet all the following criteria:
-
- They are responsible for conducting the clinical trial.
- They have access to and control over the data
- They have the right to publish the results of the trial
- They can meet all the requirements for the submission of clinical trial information.

- A ClinicalTrials.gov staff member will review the study record after it is submitted and before it is published on ClinicalTrials.gov.
- The PRS staff review process may take about 10 business days. Ensuring that the record is consistent with the ClinicalTrials.gov Protocol Review Criteria (PDF) before releasing it will expedite publication on the site.
- After it is accepted by review staff for publication, the record, including its NCT Number, will be available on ClinicalTrials.gov within 2–5 business days.
The record owner can be the PI or a study coordinator who has been delegated to this role. As Record Owner you are responsible for:
- Keeping the record up to date
- Update the record at least every 12 months until the study status is changed to “Completed”, “Terminated”, or “Withdrawn” (update at least every 6 months while actively recruiting).
- Addressing problems, errors, and major issue comments in the record in a timely manner.
- Reporting study results, if required, within 12 months of the study Primary Completion Date (usually the last study visit date).
Required Registration Updates
See related FAQ on the ClinicalTrials.gov related webpage
Responsible Parties should update their records within 30 days of a change to any of the following:
- Recruitment Status (1) and Overall Recruitment Status data elements on ClinicalTrials.gov
- Completion Date (See Primary Completion Date data element on ClinicalTrials.gov)
Other changes or updates to the record, such as protocol amendments, must be made at least every 12 months.
**It is recommended that the Record Verification Date be updated at least every 6 months for studies that are not yet completed, even if there were no changes to the record.
Results Reporting:
- The Food and Drug Administration Amendments Act (FDAAA), National Institutes of Health (NIH) require the publication of results for certain studies on ClinicalTrials.gov (Ct.gov).
- See FDAAA 801 and the Final Rule for more information.
- See How to Submit Your Results for details.
- The ICMJE Policy recommends results publication but it is not required.
- After a clinical trial has been registered on ClinicalTrials.gov and the study is completed, the Responsible Party must publish the results on ClinicalTrials.gov.
- Submission of results information is required no later than 12 months after the Primary Completion Date (the last subject last visit) of the clinical trial, which is defined as the date of final data collection for the primary outcome measure.
1 - If Overall Recruitment Status is changed to "suspended," "terminated," or "withdrawn," the Why Study Stopped data element must be submitted at the time the update is made.
If needed submit an Amendment for the following
- For studies that meet the criteria for clinical trials per NIH Definition and Applicable Clinical Trials, ensure the ClinicalTrials.gov statement is included in the Consent Form / Study Information Sheet
- Under “Study Scope” in Zot IRB, respond "yes" to the question "Does this research meet the definition of a clinical trial that requires adherence to Clinicaltrials.gov (CT.gov)?"
- Once obtained, NCT# is added to this section.
Results must be posted within 12 months of the Primary Completion Date, that is, within 12 months of the last study visit where you collected data for your primary outcome measure. This 12 month deadline has nothing to do with IRB closure, publication, data analysis, etc. so be prepared to enter your data within 12 months of your last study visit.
- Good Cause Extension
- A Good Cause Extension (GCE) request form that allows PIs to request an extension on reporting results. This form must be filled out BEFORE the one-year mark of the primary completion date. This request will not be accepted if it is past the one-year primary completion date. This form can take up to 45 days for the PRS Administrators to review. If the PI disagrees with the ruling, they will have 30 days to appeal the ruling.
- When the investigator opens up their study to the Record Summary Page, they can scroll down to the Results Section where there will be a “Delay Results” button to select. Once selected, it will take the investigator to the form they can fill out explaining why the results may be delayed.
- This document explains the process for submitting a good cause extension (GCE) request to extend the deadline for submitting clinical trial results information to ClinicalTrials.gov and describe the information necessary for the National Institutes of Health (NIH) to evaluate a GCE request and make a determination.
- Late Results Reporting Letter
- If a PI or study receives a late results reporting letter from the NIH or FDA, the PI has 30 days to post the results. Failure to do so within those 30 days will lead to enforcement action by the NIH which for an ACT includes notifying the FDA and FAPIIS (Federal Awardee Performance and Integrity Information System) as well as withholding funds from the entire institution, not just the PI.
- Yes, Federal regulations require registration and results reporting on ClinicalTrials.gov for applicable clinical trials (ACTs) under FDAAA 801 and / or per NIH Policy on the Dissemination of NIH-Funded Clinical Trial Information. The risk of noncompliance with these requirements includes civil monetary penalties in excess of $15,000 per day, civil or criminal actions, and/or loss of NIH funding for the institution.
- In relation to federally funded studies, section 402(j)(5)(A) of the PHS Act provides for the withholding of remaining or future grant funds from a grantee for failure to submit clinical trial registration and results information. ICMJE policy requires, and recommends that all medical journal editors require, registration of clinical trials prior to the start of enrollment as a condition of consideration for publication.
- Additionally, ICMJE expects authors to ensure that they have met the requirements of their funding and regulatory agencies regarding posting of results to ClinicalTrials.gov.
Why Do I Need to Register My Trial and Submit Results to ClinicalTrials.gov? Here are just some of the reasons…
- Required by Law: Section 801 of the Food and Drug Administration Amendments Act (FDAAA 801) requires responsible parties to register clinical trials and submit summary results to ClinicalTrials.gov.
- Required for Journal Publication: The International Committee of Medical Journal Editors (ICMJE) requires trial registration as a condition of the publication of research results generated by a clinical trial.
- All NIH-funded clinical trials are expected to register and submit results information to Clinicaltrials.gov, as per the "NIH Policy on Dissemination of NIH-Funded Clinical Trial Information" for competing applications and contract proposals.
- WMA Declaration of Helsinki - Ethical Principles for Medical Research Involving Human Subjects: "Every research study involving human subjects must be registered in a publicly accessible database before recruitment of the first subject" (para. 35).
What is the penalty for non-compliance?

Under NIH Policy: Noncompliance with the terms and conditions of the NIH award may provide a basis for enforcement actions, such as withholding current and future funding [45 CFR 75.371, 42 CFR 11.66]
Under Final Rule, responsible parties could be held accountable for noncompliance, with the potential for substantial civil monetary penalties, the withholding of grant funding from HHS agencies, and criminal proceedings.[2016 NEJM article]
- Case in point:
On 28 April 2021, the FDA issued its first Notice of Noncompliance to Georgia-based Accuitis who failed to submit required summary results information. The company could be “subject to a civil monetary penalty of $10,000 for each day of the violation” until the noncompliance is corrected. - FDA has issued already issued multiple Notices of Non-Compliance
- In addition, as of 12/04/2023 the FDA began publicly posting Pre-Notices for Potential Noncompliance
- This means that even if the responsible party resolves it so a Notice of Noncompliance is never sent or posted, the Pre-Notice for Potential Noncompliance and the responsible party will be publicly posted!
ICMJE: authors failing to prospectively register a trial risk its inadmissibility to journals following the ICMJE’s trial registration policy.
If the Responsible Party has joined / will leave UCI, PRS and UCI EQUIP Staff can assist in transferring a record to another PRS account. Records are registered under their sponsoring organization's account. Therefore, a sponsored study must stay in the account of its sponsor even if/when an associated investigator leaves that institution.
A UCI PRS Administrator will request the transfer by emailing PRS Staff at register@clinicaltrials.gov. To request a PRS record transfer, please email the EQUIP Manager Anu Mathur at anuradhm@uci.edu, or the EQUIP Senior Analyst Laura Ulloa at ulloal@uci.edu.
Transfers must be coordinated between the organizations involved. The receiving organization PRS Administrators are responsible for coordinating the transfer. For organizations without Administrators, the Responsible Party and Record Owners must coordinate the transfer.
To complete a record transfer, PRS Staff must receive:
- Confirmation from the receiving organization or Responsible Party that the record will be accepted (a copy of email confirmation is acceptable)
- Name of the receiving organization
- Username of the new Record Owner
- NCT number of the record
Note: Records in a Released state cannot be transferred.