How To Register and Update Your Study
The following is a quick start users guide on how to carry out some of the most common functions on ClinicalTrials.gov when registering a study. For a general overview of the registration process and requirements, see the ClinicalTrials.gov page How to Register Your Study.
Want this information in an EQUIP Tips document?
Checklist to Address Common PRS Errors
Once you have determined that your study needs to be registered on ClinicalTrials.gov, you will need to submit the record for the study in the Protocol Registration and Results System (PRS):
**PRS guided tutorials can be found here**
- For PRS access (if you do not already have an account), please contact Electronic Research Administration: era@research.uci.edu.
- If you are unsure if your study should be registered, contact your PRS Administrator:
- For UCI Health Science studies, please contact Jinah Chang (jinahec@hs.uci.edu )
- For UCI Cancer Center studies, please contact Michelle Tran (mdich@hs.uci.edu)
- For Non-UCI Health studies, please contact the EQUIP team (EQUIP@uci.edu).
- Once the account has been created, an email will be sent from ClinicalTrials.gov to the new user with a username, password and instructions for logging in to UCI’s Institutional PRS account
- For questions about the registration process at UCI, contact the EQUIP team (EQUIP@uci.edu), or contact PRS staff at register@clinicaltrials.gov.
- Go to https://register.clinicaltrials.gov/ to sign in
- Enter Organization: UCaliforniaIrvine
- Enter the username and password emailed to you by ClinicalTrials.gov

- Change your password once you log in for the first time
- Go to Accounts > Change password
- You will receive a temporary password via email
- Suggestion: Print or save a copy of the Protocol Data Entry Protocol Review Criteria (PDF) for reference. This document provides general guidance for compliant record creation as well as “Hints” detailing specific requirements for completion of key fields in the protocol record.
- Under “Quick Links”, select New Record or use the quick links to the left-hand side and click on “New Record”:

- Complete the “Create New Record” initiation screen
- Only one Record Owner can be assigned to a study record, but the Record Owner can allow other users to edit the study record by granting them access.
- For UCI Investigator Initiated studies, the Principal Investigator (Lead Researcher) or the responsible Clinical Research Coordinator can be listed as the “Record Owner”.
- Other study staff requiring PRS access should be listed in the “Access List”.

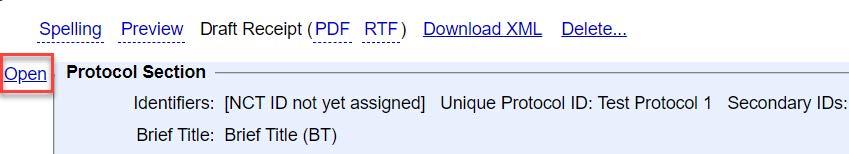
Click the Open button for the “Protocol Section"
**GENERAL EQUIP TIPS TO AVOID COMMON PRS ERRORS:
STUDY DESCRIPTION
- Brief Summary does not unnecessarily duplicate information provided for other data elements
- Brief Summary clearly states the study’s hypothesis or the purpose (for interventional and observational)
- Brief Summary and Detailed Description are written in complete sentences with no formatting errors
- Record does not use personal pronouns:“I, we, our, us, they, them, their” – becomes “the investigator(s)”; “you, your” – becomes “the participant(s)”
CONDITIONS
- Conditions/Focus of study are discrete and does not use verbs or complete sentences
- Keywords are not numbered or bulleted, each condition and keyword is listed individually, one per line
STUDY DESIGN
- All required fields are completed
- Verify Study Design based on protocol in IRB
- “Allocation” marked as “N/A” for single-arm studies
- Enrollment number Actual/Anticipated verified
ARMS/INTERVENTIONS
- Arm Title or Group/Cohort Label is brief and informative (even if there is only 1 arm)
- Interventions and intervention descriptions are entered correctly
- Arms/interventions are cross-referenced appropriately
OUTCOME MEASURES
- Title is specific and states WHAT is being measured
- only 1 variable must be assessed per outcome measure
- Description explains HOW outcome is being measured, not WHY it is being measured
- Scoring scale name, score range, significance of upper and lower limits specified (if applicable)
- Unit of measure specified
Time frame specified as a single time point or change between 2 time points
INCORRECT: “Safety and Toxicity”, Description: “Safety of study drug.”
CORRECT: “Safety as assessed by number of participants experiencing adverse events” Description: “Number of participants experiencing adverse events grade 3 or higher, as defined by Common Terminology Criteria for Adverse Events version 5.0 (CTCAE v5.0)”
ELIGIBILITY
- Age Limits are consistent with the Eligibility Criteria and with other parts of the record
- Eligibility criteria is divided into Inclusion/Exclusion criteria in bulleted format
CONTACTS/LOCATIONS
- Central Contact Person listed as a primary research team contact
- Study Officials: Person responsible for overall scientific leadership of the protocol, including the study Principal Investigator.
- Organization Affiliation: Full name of the Official’s organization (for UCI Researchers, its “University of California Irvine”)
- All study sites specified matches IRB
- Recruiting status for each study site accurate (if at least one study site is recruiting then Study Status reflects “Recruiting”)
- Each facility is listed in a separate field
IPD Sharing Statement
- Field is completed with a ‘Yes’ or ‘No’ selection
- If ‘Yes’ is selected, an IPD Sharing Plan is identified
- The Plan to Share IPD selection is consistent with the IPD Sharing Plan Description.
REFERENCES
- Each citation is listed in a separate field (if applicable)
- You will be prompted for Organization’s Unique Protocol ID.
- At UCI, the Unique Protocol ID MUST UCI IRB number (DIGITS ONLY no dashes, no #, same number as in KRP).
- Add the brief and official title for your study
- if using any acronyms- specify in the “Acronym” section
- Select Continue to complete the “Create New Record” module, and add information into each module of the protocol record as appropriate to your study.
- Secondary IDs:
- Grant‐funded projects MUST enter the sponsor‐issued grant or award number in this field. [1]
- For industry‐funded projects, use the sponsor’s protocol ID number
- For UCI Cancer Center studies, in the Secondary ID field, select "Other Identifier" and add the unique CFCCC identifier, with "Issuing Organization" as "UCI CFCCC".
- To ensure compliance with the requirements for each data entry field and module, refer frequently to the Definitions and Help links at the top of each module.
- Under the “Help” menu, go to the QUICK START GUIDE.
- Instructional links embedded throughout the PRS will aid users with navigation and record completion.
[1] U.S. National Institutes of Health (NIH) Grant/Contract Award Number: In the Secondary ID field, include activity code, institute code, and 6-digit serial number. Other components of the full award number (type code, support year, and suffix) are optional.
**EQUIP TIPS TO AVOID COMMON PRS ERRORS:
- Unique protocol ID is the IRB number ONLY (DIGITS only, no dashes, letters or #)
- Brief Title does not include study type (e.g., Phase I, Randomized…)
- Secondary IDs include NIH grant #s (verify in IRB Record) or CFCCC studies: Include additional a unique identifier
- The Record Verification Date reflects the last time the PRS record was updated. Revise this date each time the record is verified for accuracy and completeness.
- Complete the rest of the required information per guidance in this section:

- If unsure of dates, indicate the date as “anticipated” in the last dropdown under “Type”
- If recruitment has begun, specify the “actual” date the first subject was enrolled.
**EQUIP TIPS TO AVOID COMMON PRS ERRORS:
- Record Verification Date is the current month/year
- Overall Status matches IRB Status (in KRP)
- Completion Dates Actual/Anticipated have been evaluated for accuracy
- If timeframes for outcomes are the same, the primary and study completion dates are identical
- For all UCI Investigator Initiated studies, the Principal Investigator (Lead Researcher) should be designated as the Responsible Party (RP).
- Under Responsible Party choose “Principal Investigator” (PI)
- Select the Investigator name from the drop-down menu (email era@uci.edu if the PI does not have a PRS account)
- Enter the Investigator’s Official Title
- Investigator’s Affiliation should automatically populate to “University of California, Irvine”
- Sponsor: Regardless of funding source, enter the “regulatory sponsor” (primary organization overseeing the implementation of the study), usually University of California Irvine
- Any collaborating sites should be entered under the Collaborators section
- Click Continue
- U.S. FDA Regulated Drug / Device: Indicate whether this study involves an FDA‐regulated drug, biologic, or device.
- U.S. FDA Regulated Drug / Device: Indicate whether this study will be conducted with a drug/device product under a U.S. FDA Investigational New Drug (IND) Application or Investigational Device Exemption (IDE).
- Human Subjects Protection Review: Enter the current status of your IRB Protocol in KRP (e.g., “submitted, pending” if an application has been submitted but is under review.
- Add the following information for the UCI IRB:
- Board Name: University of California Irvine IRB
- Board Affiliation: University of California Irvine
- Board Contact: 949-824-8170
- Email: irb@uci.edu
- Business Address: 160 Aldrich Hall, Irvine, California, 92697-7600
- Data Monitoring: Indicate whether a data monitoring committee (board) has been appointed for this study
- Oversight Authorities: Name each national or international organization with authority over the protocol (e.g. DHHS, FDA, NIH, DOD, DOE, etc.)
**IMPORTANT: For studies that meet the criteria for clinical trials per NIH Definition and Applicable Clinical Trials, Consent / study information sheet contains requisite ct.gov language
**EQUIP TIP TO AVOID COMMON PRS ERRORS:
- IND/IDE information completed (if applicable)
- Enter all the applicable information regarding the study you are registering in the Study Description”, “Conditions”, “Study Design”, “Arms and Interventions”, “Eligibility” sections, as well as any other applicable sections.
- Review and reference the PRS User Guide
**Tip: Data is saved as each page is completed so that you can "Quit" at any time, saving the record to come back to and submit later**

- After filling in the last data entry page, the “Protocol Section” page appears with all of the information Review and “Entry Complete” if all information is complete.
- Address any ERROR messages if populated.
**GENERAL EQUIP TIPS TO AVOID COMMON PRS ERRORS:
- RECORD VERIFICATION DATE is current Month and year
- Unique Protocol ID is the IRB number ONLY (No other Unique protocol ID’s, IRB number in digits only, no dashes, #)
- Record Owner is the PI or Research Coordinator
- Contact info for Record Owner is up-to-date
- PI on record matches IRB PI
- NCT# included in IRB “Clinical Trials Information” section
- All Warnings/Errors addressed
- All parenthetical citations have been removed
- All acronyms have been expanded on their first use
- Spell-check complete
- Free-text fields are blank if there is no information to report, and do not contain text such as “TBD,” “Pending,” “N/A,” “None”
- If the PI is the responsible party, they will review all entries made by study staff prior to release.
- Once the information is confirmed (passed internal review), under “Record Status” – click “Approve”:

- Once the information is confirmed (passed internal review), under “Record Status” – click “Approve”:
- Select “Release” to submit the record to ClinicalTrials.gov. The responsible party confirms their identity, and that the record is up to date, reviewed for accuracy and completeness:
- Once released, PRS staff perform final review and processing of the record.

- Note: some records may receive PRS Review Comments that identify major issues that must be addressed by the responsible party before the record will be made available on ClinicalTrials.gov.
- Following successful PRS review, records are made available to the public through the ClinicalTrials.gov web site within 2 to 5 days of Release.
- The ClinicalTrials.gov Identifier (NCT number) is assigned as part of that process.
- Please download and review the IRB EQUIP TIPS guidance document called “CHECKLIST TO ADDRESS COMMON PRS ERRORS” to avoid common issues noted by the ClinicalTrials.gov database (PRS) staff prior to public release.
- Directly email PRS staff if you have any questions or need help related to PRS Review Comments at register@clinicaltrials.gov
- Once a record is created, you can update it as long as it is not undergoing PRS review:
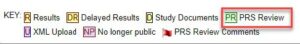
- If your department is utilizing the “Secondary ID” Section for a Unique department ID, click “Show/Hide Columns” next to the search bar in PRS, and select “Secondary IDs” as part of your records filter:
When / how often must I update ClinicalTrials.gov registration information?
- The Record Verification Date should be updated any time the responsible party reviews the complete set of submitted clinical trial information for accuracy and not less than every 12 months, even if no other updated information is submitted at that time.
- Responsible Parties should review the Clinical Trial Registration Data Elements for More Frequent Updating Table as many elements must be updated within 30 days of a change.
- It is recommended that the Record Verification Date be updated at least every 6 months for studies that are not yet completed, even if there were no changes to the record.
Clinical Trial Registration Data Elements for More Frequent Updating
| Data Element | Deadline for Updating (i.e., not later than the specified date) |
|---|---|
| Study Start Date | 30 calendar days after the first subject is enrolled (if the first human subject was not enrolled at the time of registration). |
| Intervention Name(s) | 30 calendar days after a nonproprietary name is established. |
| Availability of Expanded Access | 30 calendar days after expanded access becomes available (if available after registration); and 30 calendar days after an NCT number is assigned to a newly created expanded access record. [1] |
| Expanded Access Status | 30 calendar days after a change in the availability of expanded access. |
| Expanded Access Type | 30 calendar days after a change in the type(s) of available expanded access. |
| Overall Recruitment Status | 30 calendar days after a change in overall recruitment status. [2] |
| Individual Site Status | 30 calendar days after a change in status of any individual site. |
| Human Subjects Protection Review Board Status | 30 calendar days after a change in status. |
| Primary Completion Date | 30 calendar days after the clinical trial reaches its actual primary completion date. |
| Enrollment | At the time the primary completion date is changed to "actual," the actual number of participants enrolled must be submitted. |
| Study Completion Date | 30 calendar days after the clinical trial reaches its actual study completion date. |
| Responsible Party, by Official Title | 30 calendar days after a change in the responsible party or the official title of the responsible party. |
| Responsible Party Contact Information | 30 calendar days after a change in the responsible party or the contact information for the responsible party. |
| Device Product Not Approved or Cleared by U.S. FDA | 15 calendar days after a change in approval or clearance status has occurred. |
| Record Verification Date | Any time the responsible party reviews the complete set of submitted clinical trial information for accuracy and not less than every 12 months, even if no other updated information is submitted at that time. |
- The Food and Drug Administration Amendments Act (FDAAA), National Institutes of Health (NIH) require the publication of results for certain studies on ClinicalTrials.gov (Ct.gov).
- See FDAAA 801 and the Final Rulefor more information.
- See How to Submit Your Results for details.
- The ICMJE Policy recommends results publication but it is not required.
- After a clinical trial has been registered on ClinicalTrials.gov and the study is completed, the Responsible Party must publish the results on ClinicalTrials.gov.
- Submission of results information is required no later than 12 months after the Primary Completion Date (the last subject last visit) of the clinical trial, which is defined as the date of final data collection for the primary outcome measure.
PARTICIPANT FLOW
- Protocol Enrollment refers to total number of subjects who consented to protocol (including screen failures, withdrawals, etc.)
- Recruitment details (optional) explains any specifics used at time of recruitment
- Pre-assignment details explains (in detail) what happened to subjects who signed consent but were not assigned to an arm/intervention (i.e., how many screen failures, withdrawals, etc.)
- Arms and arm descriptions specified consistent with protocol section
- Number of Participants Started refers to total number of participants assigned to each arm
- Number of Participants Completed refers to total number of participants who completed study intervention
- Reason(s) for Not Completed provided
- Divided into periods/milestones appropriately
- Total number of participants started cannot be greater than enrollment number
BASELINE CHARACTERISTICS
- Overall Number of Baseline Participants should match Number of participants Started (from Participant Flow)
- Baseline Analysis Population Description explains if there is a discrepancy between Overall & Started numbers
- Arm titles/descriptions are consistent with participant flow and/or protocol section
- Data is presented per arm
- If “number of participants” is reported, make sure Measure Type is “Count of Participants”
- Measure description is specified for all Study-specific measures
OUTCOME MEASURES
- Titles/descriptions/time frame meet the criteria (as specified on prior checklist)
- Results are reported per arm
- Population Analysis Description includes reason why Number of Participants analyzed is different than total number of participants completed (if applicable)
- Type and Number of Units analyzed is indicated, if other than “number of participants” (i.e., # of Lesions)
- Unit of measure matches what is stated in Outcome Title/Description
- Sum of all results entered for each arm equals overall number of participants analyzed
- Verify true data is entered and there are no placeholders
- Statistical Analysis portion is completed
ADVERSE EVENTS
- Time frame specified
- Collection Approach specified
- Arm titles/descriptions consistent with other sections in the record
- Data presented per arm
- All-cause mortality specified (cross-check with number “not completed due to death” from participant flow and any mortality measures in outcome section, if applicable)
- Total Number “At Risk” must be equal to total number of participants who started the study
CERTAIN AGREEMENTS
- Disclosure restrictions should be ‘No’ unless documentation is presented to the contrary
RESULTS POINT OF CONTACT
- Responsible Party's Contact information will be public facing
- Information is correct and valid email address/phone number entered
DOCUMENT SECTION
- Documents in pdf/a format
- Protocol (required for primary completion date after January 18, 2017)
- Statistical Plan (required for primary completion date after January 18, 2017)
- Informed Consent Form (required for studies approved on or after January 21, 2019)
- Cover Page
-Record (NCT) Number
-Study Title
-PI Name
-Date of Document (must match date within actual document)
REFERENCES
- Links are verified (if applicable)
- WHICH STUDIES?
- All clinical trials funded by an agency that has signed onto the Common Rule initially approved on or after January 21, 2019 OR that transitioned to the 2018 Common Rule Requirements (45 CFR 46.101(l)) and the transition determination was documented and dated by the IRB before the trial is closed to recruitment and 60 or fewer days before the last protocol-required study visit by any subject enrolled in the protocol.
- Please see OHRP “The Informed Consent Posting Requirement” PowerPoint for details.
- WHAT DO I POST?
- One IRB-approved consent form used to enroll subjects
- Even if multiple versions were approved by the IRB, only one must be uploaded.
- WHEN?
- After the clinical trial is closed to recruitment, and no later than 60 days after the last study visit by any subject, as required by the protocol.
- WHERE?
- Two federal websites have been identified as satisfying 45 CFR46.116(h): gov and a designated docket folder on Regulations.gov (Docket ID: HHS-OPHS-2018-0021).
- HOW?